cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;cro企业,cro公司,cro,北京cro,北京cro公司,北京cro企业;

20150206《药物临床试验质量管理规范》修订稿(征求意见稿)起草说明
2022年03月28日发布


《药物临床试验质量管理规范》修订稿
(征求意见稿)起草说明
国家食品药品监督管理总局于2021年启动《药物临床试验质量管理规范》(简称“GCP”)修订,现完成修订稿起草,有关情况说明如下:
一、修订背景
现行GCP自2003年9月1日施行以来,有力地促进了药物临床试验质量的提高,加强了受试者权益与安全保障。随着药物研发形势的发展,在GCP实施过程中出现一些与新形势新要求不相适应之处。因此根据药物研发及其监管的实践需求,结合发展趋势,对部分条款予以修改。
二、修订经过
食品药品监管总局于2021年将GCP修订列入立法计划。2021年7月前,药化注册司组织分析了现行GCP中存在的与现状不适用的内容,并与国外相关法规进行了比对研究。
2021年7月,药化注册司会同国家卫生计生委医政医管局在长沙召开会议,与总局核查中心以及部分省局监管人员、药物临床试验机构与研发企业代表,就GCP修改进行了初步研讨和征求意见。
根据前期的分析研究与意见收集,药化注册司组织形成GCP修订意见草案,分别于2021年9月、10月,在天津和北京召开意见征求会,与总局法制司、核查中心、药审中心以及部分省局监管人员、药物临床试验机构、伦理委员会以及研发企业代表等,就GCP修订意见进行讨论和修改,形成征求意见稿。
三、修订内容与说明
根据实践需求以及发展趋势,仅对部分条款进行必要修改。保持原章节结构不变,修改条文23条,新增条文11条,着重修订以下几方面内容:
(一)强调申办者的职责,落实主体责任。明确由申办者负责制定试验方案(第16条),补充了试验方案应包含的必要内容(第17条),要求申办者评估拟委托的第三方并对其工作质量负责(新增第8条);要求申办者负责对药物临床试验安全性进行持续评估,对严重不良事件进行分析评估后分类报告(第40条)。
(二)提高伦理审查工作规范性,加强受试者保护。明确哪些文件必须经伦理委员会批准后方可执行(第10条),补充了伦理审查的重点内容(第12条),强调伦理委员会应对试验项目进行定期审查(新增第4条),加强对弱势群体以及健康受试者的保护(新增第3条);鼓励伦理委员会与技术审评同步开展伦理审查(新增第2条),以期缩短审批时间,促进伦理审查能力和水平的提高;强调了药物临床试验受试者保护是临床试验各参与方共同的职责(第8条),补充了知情同意时需要告知受试者的必要内容并规范知情同意过程(第14、15、24条)。
(三)引入对药物临床试验机构的职责要求,加强机构对药物临床试验的管理。在强调研究者责任的落实的同时,要求药物临床试验机构建立与临床试验管理相适应的管理体系,对临床试验进行管理(新增第5条);要求申办者向药物临床试验机构通报临床试验中的有关信息(第41、44、47条)。
(四)加强省局在信息报告与备案管理中的作用,落实临床试验日常监管。要求伦理委员会向所在地省局备案并报告年度伦理审查情况(第9条);要求研究者及申办者及时向所在地监管部门报告药物临床试验中所发生的严重不良事件(第26、40条);要求药物临床试验机构向所在地省级食品药品监督管理部门报告年度药物临床试验管理情况(新增第7条)。
(五)要求药物临床试验有关信息公开,减少利益冲突,强化社会监督。要求申办者落实药物临床试验登记与信息公示(新增第9条);规定伦理委员会应确保工作的独立性并公开必要的信息,伦理委员应签署利益冲突声明(新增第1条);要求研究者主动声明和公开任何与临床试验相关的利益冲突情况(第29条)。
(六)优化药物临床试验严重不良事件(SAE)报告流程,明确报告要求。明确申办者应负责试验药物风险/收益的持续评估(第40条)。在报告流程方面,要求研究者将本机构发生SAE立即报告责任主体(申办者),报告“日常管理部门”(伦理委员会及所在地省级监管部门),而不必再重复报告总局(第26条);要求申办者负责对SAE等安全性相关信息进行分析评估,按照不同分类的时限要求向总局药审中心进行个案报告和年度报告(第40条)。
(七)提出生物等效性试验用药品留样要求,加强仿制药临床试验管理。要求申办者提供给临床试验机构和研究者足够数量的试验用药品以供随机抽取;要求申办者委托临床试验机构保存试验用药品留样或由机构委托独立的第三方保存留样备查,留样不得返还申办者或与其利益相关的第三方。保存时限为药品上市后至少2年。(新增第11条)
另,与《药品注册管理办法》第30条一致,对GCP第3条进行修改,规定药物的临床试验(包括人体生物利用度或生物等效性试验)须遵照GCP;鉴于《世界医学大会赫尔辛基宣言》版本不断更新,对GCP第4条进行修改,不再附《世界医学大会赫尔辛基宣言》内容;针对当前临床试验电子数据管理系统越来越普及的现状,新增第10条提出电子数据管理系统应经过验证、数据修改应留有稽查痕迹等要求;还对个别错别字以及不确切之处进行了修改。

地址:北京市丰台区西四环南路35号中都科技大厦10层
《互联网药品信息服务资格证书》 编号:(京)一非经营性一2021一0014
备案序号:京ICP备12047146号和京ICP备16030590号 信息产业部备案管理系统网址

扫
一
扫
© 2026北京远博康惠医药信息咨询有限公司 版权所有
北京远博医药CRO
感谢您关注远博医药微信公众号


© 2026北京远博恒康医药科技有限公司 版权所有

商务拓展部(药品&器械)袁经理
电话:010-
邮箱:ybkj045@126.com
人力资源部主任
电话:13146546661
邮箱:ybkj010@126.com
商务总监(药品&器械)薛总
电话:13810708945
邮箱:ybkj027@126.com








◆丰富的临床试验服务经验:
◆熟悉政策法规及审评动态:
◆良好的专家关系:
◆大量临床医院的资源:
◆专业的数据管理:
CRO:Contract Research Organization, 出现于上世纪80年代,一种学术性或商业性的科学机构。申办者可委托其执行临床试验中的某些工作和任务,此种委托必须作出书面规定,其目的是通过合同形式向制药企业提供新药临床服务的专业公司。CRO 可以作为制药企业的一种可借用的外部资源,可在短时间内迅速组织起一个具有高度专业化的和具有丰富临床经验的临床队伍,并能降低整个制药企业的管理费用。
 了解更多了解更多
了解更多了解更多

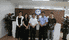






我公司承接国内外厂家或公司的委托,从事药物、医疗器械临床代理服务,以下是我公司服务过的部分项目的名称。














北京远博医药CRO成立于2005年,为医疗企业提供临床试验、项目申报、项目管理和信息咨询等的专业化外包服务(CRO服务),主要为专业化服务内容包括:新药(化药、中药和生物制品)的Ⅰ~Ⅳ临床试验、药代动力学(PK)、生物等效性(BE)、医疗器械的临床试验、上市后再评价、中药保护临床试验、药物经济学等项目的监查及项目管理。
专业化人才优势
绝大多数员工拥有专科以上学历
高标准质量管理体系
必须通过国家药监局高级研修学院的GCP培训并获得证书
完善的培训系统
注重对员工的技术培训,积极参加国家药监总局、药审中心、器械审评中心及各学术团体举办的专业培训及会议;
◆丰富的临床试验服务经验:
◆熟悉政策法规及审评动态:
◆良好的专家关系:
◆大量临床医院的资源:
◆专业的数据管理:
◆优秀而稳定的团队:
◆完善的标准操作流程:
◆独立的质量保障部门:
◆灵活多样的服务方式:






































































































 预约上门洽谈免费项目咨询
预约上门洽谈免费项目咨询CRO

北京远博医药

临床试验、一致性评价、BE研究和技术服务的专家
专业为医药企业提供一体化的专业技术服务


临床试验优先选择的CRO
18618362640
010-
全国客服热线